A Two Bit Decoder
The entire human genome is available as a single .2bit file
here (click on
“Full Data Set”, then download hg19.2bit). Unlike the stellar signal
in His Master’s Voice, the 2bit
format is reasonably
clearly documented.
We want to write Clojure code to:
-
Provide base pairs in symbolic (rather than raw binary) form as lazy sequences – i.e., sequences which need not all fit in memory at once, but can be consumed and processed as needed;
-
Provide “random access” to this data selectively, e.g. by chromosome, rather than always reading through the entire file;
-
Provide access to metadata encoded in the file.
The functionality to do this is posted in the jenome project on GitHub. In this post, we’ll explore this code a little; in following posts, we’ll do some investigating of the actual genome per se.
Capability (2) is provided, in part, by implementing random-access
reads from file fname of len bytes starting at offset:
(ns jenome.rafile
(:import (java.io RandomAccessFile)))
(defn read-with-offset [fname offset len]
(let [raf (RandomAccessFile. fname "r")
bb (byte-array len)]
(.seek raf offset)
(.readFully raf bb)
(.close raf)
bb))Armed with this, we can get the .2bit file header:
(defn file-header [fname]
(let [[sig ver seqcnt resvd] (->> (read-with-offset fname 0 16)
(partition 4)
(map bytes-to-number))]
(assert (= sig 0x1A412743))
(assert (= ver 0))
(assert (= resvd 0))
seqcnt))file-header basically just gives us the number of sequences
(usually, chromosomes) in the file, doing some sanity checks along the
way. bytes-to-number converts an arbitrary sequence of bytes to the
appropriate unsigned integer. (For brevity’s sake, I won’t show every
utility function in this blog post; the source code on
GitHub is reasonably short.)
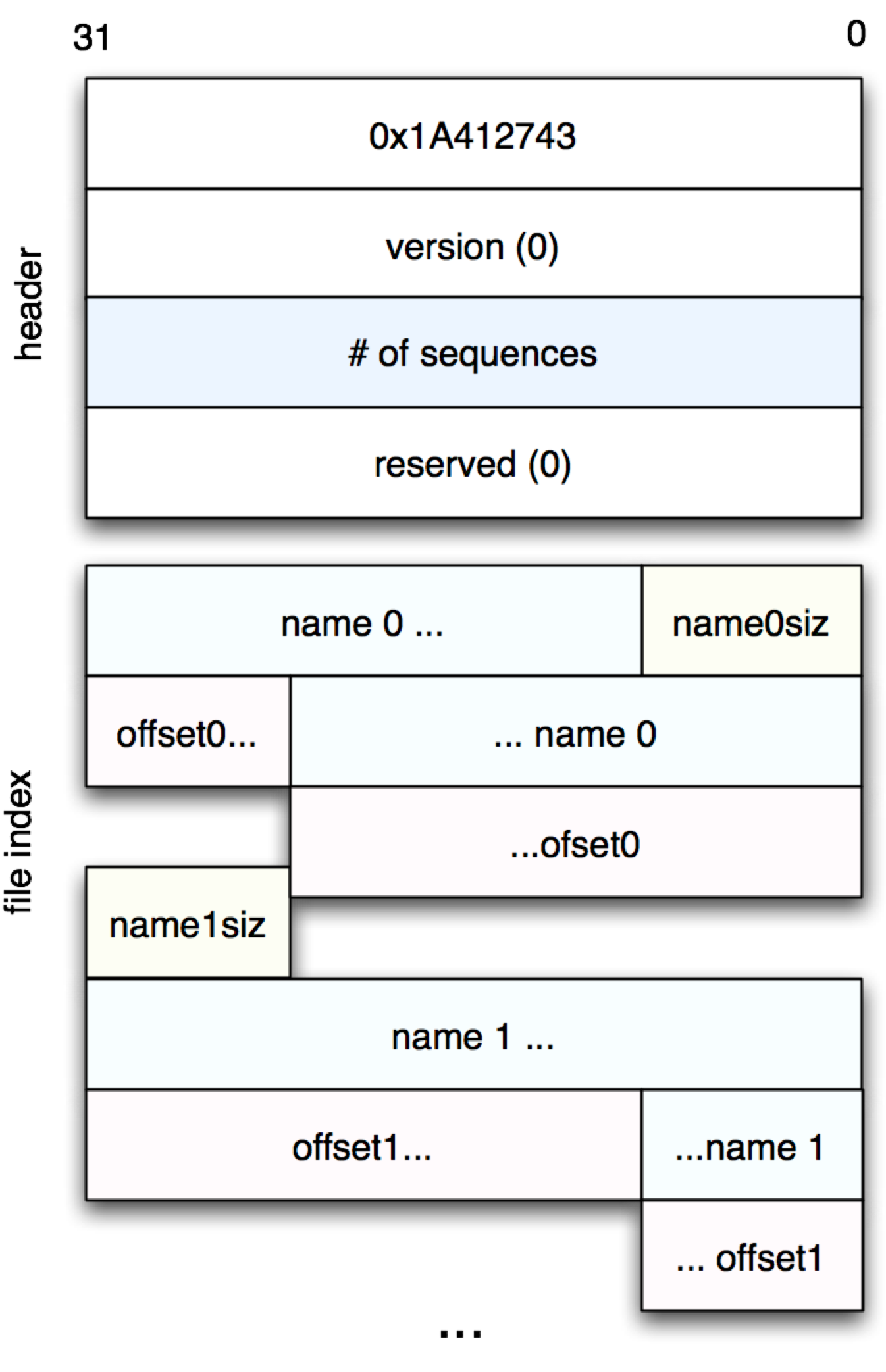
Figure 1: Header and File Index
The next part of the file, as shown in Figure 1, is called the “file index,” and contains a list of sequences contained the rest of the file. It can be read as follows:
(defn file-index [fname seqcnt]
(loop [i 0
ofs 16
ret []]
(if (< i seqcnt)
(let [[nlen] (read-with-offset fname ofs 1)
name (apply str (map char (read-with-offset fname
(+ ofs 1) nlen)))
seq-offset (get32 fname (+ ofs 1 nlen))]
(recur (inc i) (+ ofs nlen 5) (conj ret [nlen name seq-offset])))
ret)))This somewhat imperative code walks through the seqcnt sequence
portions of the index, pulling out sequence names and lengths as we
go.
It’s here that we introduce a new friend, the yeast Saccharomyces
cerevisiae
(SacCer3), used since antiquity for making bread and fermented
beverages. Relatively small in comparison with the human genome,
SacCer3 will be our “unit test” organism. Available
here
and checked into resources, the file can be accessed as
(def yeast
(as-file (resource "sacCer3.2bit")))(I have imported resource and as-file from clojure.java.io; again, see the source code.)
Our index-reading code yields:
(let [seqcnt (file-header yeast)]
(file-index yeast seqcnt))
;=>
[[4 "chrI" 191]
[5 "chrII" 57762]
[6 "chrIII" 261074]
[5 "chrIV" 340245]
[5 "chrIX" 723245]
[4 "chrV" 833233]
[5 "chrVI" 977468]
[6 "chrVII" 1045025]
[7 "chrVIII" 1317776]
[4 "chrX" 1458453]
[5 "chrXI" 1644907]
[6 "chrXII" 1811627]
[7 "chrXIII" 2081188]
[6 "chrXIV" 2312312]
[5 "chrXV" 2508412]
[6 "chrXVI" 2781251]
[4 "chrM" 3018284]]The apparent consistency of these values give us some
initial confidence that we are reading the index correctly. Note,
however, the curious fact that chrIX appears between IV and V.
With this encouraging start, we can now attack the sequences proper. These are laid out as shown in Figure 2, with block metadata prepended to the actual DNA sequences:

Figure 2: Sequence Record Layout
The “N blocks” are blocks of unknown sequences with specified offsets
and lengths. Masked blocks are blocks which are known repetitions
(indicated as lower case “a”, “g”, “c” and “t” in the text-based
‘FASTA’ file format). We are obviously most interested in dnaSize,
the number of base pairs in the sequence, and the actual sequence
values themselves.
Unpacking the above data format (except the base pairs per se)
makes heavy use of get32, which just returns the unsigned 32-bit
integer at the specified file location. This code doesn’t need to be
super efficient, since the block headers themselves are quite small. The
metadata for the entire file is returned as a sequence of maps.
(defn getblk [fname offset n]
(let [ret (map #(get32 fname (+ offset (* 4 %))) (range n))
offset (skip offset n)]
[ret offset]))
(defn sequence-headers
"
Get sequence headers from .2bit file, as documented in
http://genome.ucsc.edu/FAQ/FAQformat#format7. Returns a list of maps
with details for each sequence.
"
[fname]
(let [seqcnt (file-header fname)]
(for [[nlen name ofs] (file-index fname seqcnt)]
(let [[[dna-size] ofs] (getblk fname ofs 1)
[[n-block-count] ofs] (getblk fname ofs 1)
[n-block-starts ofs] (getblk fname ofs n-block-count)
[n-block-sizes ofs] (getblk fname ofs n-block-count)
[[mask-block-count] ofs] (getblk fname ofs 1)
[mask-block-starts ofs] (getblk fname ofs mask-block-count)
[mask-block-sizes ofs] (getblk fname ofs mask-block-count)
[[reserved] ofs] (getblk fname ofs 1)]
(assert (zero? reserved))
{:name name
:nlen nlen
:dna-size dna-size
:n-block-count n-block-count
:n-block-starts n-block-starts
:n-block-sizes n-block-sizes
:mask-block-starts mask-block-starts
:mask-block-sizes mask-block-sizes
:dna-offset ofs}))))A few sanity checks are included in test_core.clj to make sure we’re
decoding the metadata correctly. Requirement (3) is done!
The final step (Requirement (1)), is to actually get our base pairs (BPs).
Since we have to assume the data set is very large (as is the case
with the human genome), we cannot read the entire DNA sequence at
once. The first part is to break the dna-size base pairs (remember
we have 2 bits per BP, or 4 BP/byte), starting at dna-offset. First
we obtain the “coordinates” of the sequences we want to read:
(defn get-buffer-starts-and-lengths
"
Return buffer offsets (starting at ofs) required to cleanly read a
total of m bytes no more than n at a time
"
[ofs n m]
(loop [a ofs
len n
ret []]
(if (>= a (+ m ofs))
ret
(recur (+ a n)
n
(conj ret [a (min len (- (+ m ofs) a))])))))
;; Example:
(get-buffer-starts-and-lengths 100 200 512)
;=> [[100 200] [300 200] [500 112]]At long last, having obtained the locations and lengths we want to read from, we can get our sequences out:
(defn genome-sequence
"
Read a specific sequence, or all sequences in a file concatenated
together; return it as a lazy seq.
"
([fname]
(let [sh (sequence-headers fname)]
(mapcat #(genome-sequence fname %1 %2)
(map :dna-offset sh)
(map :dna-size sh))))
([fname ofs dna-len]
(take dna-len
(apply concat
(let [byte-len (rounding-up-divide dna-len 4)
starts-and-lengths (get-buffer-starts-and-lengths
ofs 10000 byte-len)]
(for [[offset length] starts-and-lengths
:let [buf (read-with-offset fname offset length)]
b buf]
(byte-to-base-pairs b)))))))
;; Example:
(->> yeast
genome-sequence
(take 30))
;=> (:C :C :A :C :A :C :C :A :C :A :C :C :C :A :C
; :A :C :A :C :C :C :A :C :A :C :A :C :C :A :C)This function allows us to choose the entire genome, or that for a given offset and number of base pairs (whether from the metadata for an entire chromosome, or some smaller region, thus satisfying (2)).
The next post will focus on verification of correctness of this code; subsequent posts will begin to explore the characteristics of this data for various genomes, human or otherwise.
NEXT: Validataing the Genome Decoder
blog comments powered by Disqus