Getting Our Hands Dirty (with the Human Genome)
In the past two posts, we created and validated a decoder for genome data in 2bit format.
Let’s start actually looking at the human and yeast genomes per se. First, if you haven’t downloaded it yet, you’ll need a copy of the human genome data file:
cd /tmp
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit
(Depending on your local bandwidth conditions, this can take quite awhile.)
Again we define convenience vars for the two data files:
(def human "/tmp/hg19.2bit")
(def yeast (as-file (resource "sacCer3.2bit")))The simplest thing we can do is count base pairs:
jenome.core> (time (->> yeast genome-sequence count))
"Elapsed time: 17577.042 msecs"
12157105
jenome.core> (time (->> human genome-sequence count))
"Elapsed time: 4513537.422 msecs"
-1157806032WTF?! count has overflowed (if you convert -1157806032 to an
unsigned int you get 3137161264, which makes more sense). This, and
our running time of 75 minutes, is our first hint of pain associated
with handling with such a large dataset.
It’s worth noting that number overflows in Clojure generally raise an
ArithmeticException (or silently get promoted to BigIntegers,
depending on the operators used) – I emailed the clojure mailing
list about this
surprise and a ticket has been
made.
The overflow is easily enough remedied with a non-overflowing count
(inc' is one of the aforementioned promoting operators):
(defn count' [s] ;; Regular count overflows to negative int!
(loop [s s
n 0]
(if (seq s)
(recur (rest s)
(inc' n))
n)))Which gives us the unsigned version of our previous answer, in 10% more time:
jenome.core> (time (->> human genome-sequence count'))
"Elapsed time: 4931630.301 msecs"
3137161264
jenome.core> (float (/ 3137161264 12157105))
258.05167(Update 2/14/2014: Matthew Wampler-Doty came up with a more elegant count':
(defn count' [s] (reduce (fn [x _] (inc x)) 0 s))which is about 25% faster than my slightly more naïve version.)
We now have our first bit of insight into human vs. yeast genomes: we humans have about 260x more base pairs than yeast do.
The next question is, What are the relative proportions of occurrence
of each base pair? Easily answered by the frequencies function:
jenome.core> (->> yeast genome-sequence frequencies)
{:C 2320576, :A 3766349, :T 3753080, :G 2317100}
jenome.core> (->> human genome-sequence frequencies)
{:T 1095906163, :A 854963149, :C 592966724, :G 593325228}
jenome.core> Those numbers have a lot of digits, so here are some bar charts:
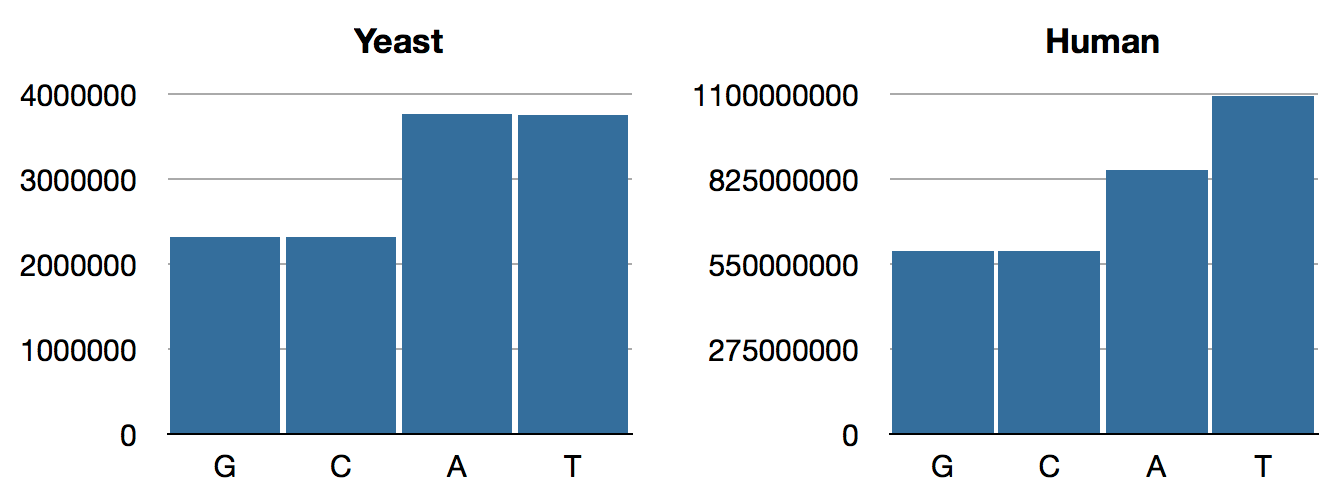
Figure 1: Our first stab at nucleotide frequencies
Though we are playing ignorant data analysts rather than real biologists, this looks a bit fishy right out of the gate. Why so many Ts?
As a sanity check, perhaps we ought to look more closely at the actual data:
jenome.core> (->>
human
genome-sequence
(map name)
(take 100)
(apply str))
;=> "TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT
; TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT"That doesn’t look good at all. How are the first 10000 base pairs distributed?
jenome.core> (->>
human
genome-sequence
(take 10000)
frequencies)
;=> {:T 10000}How about the next 10,0000?
jenome.core> (->>
human
genome-sequence
(drop 10000)
(take 10000)
frequencies)
;=> {:T 2012, :A 2065, :C 3028, :G 2895}That looks a lot better. Playing around with this shows that exactly the first 10,000 base pairs are Ts.
The astute reader will recall that the file index in our decoder had slots for unknown (“N-block”) sequences. Though we didn’t explore them explicitly, S. Cerviciae’s genome had nothing in these slots. It’s time we went back and looked at our human’s file index:
(->> human
sequence-headers
(map (juxt :name :dna-size :n-block-count))
clojure.pprint/pprint)
;; =>
(["chr1" 249250621 39]
["chr2" 243199373 24]
["chr3" 198022430 9]
["chr4" 191154276 12]
["chr5" 180915260 7]
["chr6" 171115067 11]
["chr7" 159138663 17]
["chrX" 155270560 23]
["chr8" 146364022 9]
;; LOTS more entries
)Holy cow, they all have N-blocks. This is only the first section; this Gist shows the entire set of N-blocks for all the sequences. (It also shows that there are many more files in the index than the yeast genome had, and many more than one for each of the 24 human chromosomes – clearly there are many things to learn about this data.)
Moreover, if we look at the N-blocks for the first sequence:
(->> human
sequence-headers
first
((juxt :n-block-starts :n-block-sizes))
(apply interleave)
(partition 2)
(map vec)
clojure.pprint/pprint)
;; Gives:
([0 10000]
[177417 50000]
[267719 50000]
[471368 50000]
;; ... total of 39 entries.
)As expected, our first 10,000 base pairs are actually unknown. The 2-bit file format apparently uses 0’s for the unknowns AND to represent Ts. The rest of the offsets and lengths are in this Gist. (Perhaps someone can explain to me why these are all multiples of 10000!)
All this means that our decoder needs a tweak to give us :N instead of
:T when the position of the base pair is inside one of the N-blocks.
This we will accomplish in the next post, before we proceed to the
study of other properties of the genome.
Before we end, it’s worth noting that, had we been real biologists, we would surely have known about Chargaff’s Rule, which states that A and T ratios should be the same, as are G and C, as born out in our yeast distribution, above.
Though we haven’t gained mastery over too much of the genome yet, it
is worth pointing out Clojure’s strengths in the above and previous
posts. With comparatively little code we are able to investigate many
of the features of these data sets, thanks to the power of the
threading macro ->>, higher-order functions such as juxt and map,
and sequence handling functions such as interleave,
partition, and frequencies. We’ve also processed large data sets
“lazily” without needing to fit large amounts of data in memory at
once. And we have hardly scratched the surface of Clojure’s tools for
parallel computation, though we may make more use of these in coming posts.
Next: Updating the Decoder
blog comments powered by Disqus